
Negli ultimi anni Apple ha fatto leva sull?anima Unix del suo sistema operativo MacOSX per muovere importanti passi nel settore dei server, un mercato in cui può già vantare clienti del calibro di Oracle e Cisco.
Con l’arrivo del processore G5, i sistemi di Apple sono diventati molto appetibili anche per la realizzazione di cluster di server a basso costo che, come nel noto caso del supercomputer realizzato presso la Virginia-Tech University, possono raggiungere potenze di calcolo di tutto rispetto. Altri sistemi simili sono in fase di realizzazione presso varie aziende e organizzazioni: l?esercito americano, ad esempio, ne ha commissionato uno per effettuare delle simulazioni aerodinamiche del volo a velocità ultrasonica e per compiere studi sulla fluidodinamica.
Fra le prime realtà italiane ad aver scelto una soluzione di supercomputing basata su macchine Apple c?è l’Istituto Veneto di Medicina Molecolare ( VIMM ), che sta utilizzando un cluster di Xserve G5 per realizzare simulazioni di dinamica molecolare su sistemi di grandezza comparabile ai più grandi modelli attualmente simulati a livello internazionale.
Punto Informatico ha intervistato il Dott. Sergio Pantano, responsabile del progetto, e il Sig. Pierpaolo Fantuzzi di MacAT , il tecnico hardware/software che sta seguendo la realizzazione del cluster.
Punto Informatico: Dott. Pantano, ci può spiegare cos’è il VIMM e di cosa si occupa?
Sergio Pantano: L? istituto veneto di medicina molecolare (o Venetian Institute of Molecular Medicine, VIMM) è un centro di ricerca avanzata dedicato alla investigazione basica e applicata nel campo delle scienze biologiche e mediche.
PI: Può raccontarci qualcosa di lei e del suo ruolo in questo progetto?
SP: Io sono dottore in fisica specializzato in biofisica computazionale, interazioni fra proteine e modellizzazione molecolare. Mi sono recentemente incorporato al VIMM dove lavoro in collaborazione con diversi gruppi di ricerca in biologia molecolare e cellulare. Fra diversi progetti di ricerca in bioinformatica strutturale, sto portando avanti lo studio di un grosso complesso di proteine di membrana: la pompa al Calcio (Calcio-ATPasi del reticolo sarcoendoplasmatico o SERCA, dal suo acronimo in inglese) e il Fosfolambano (PLB), una piccola proteina che modifica il funzionamento della SERCA. Queste due proteine sono molto importanti, poiché regolando la concentrazione di calcio all?interno delle cellule muscolari cardiache controllano il battito del cuore.
PI: Ci può illustrare i dettagli di questa simulazione?
SP: La struttura delle proteine di membrana è molto difficile da ottenere sperimentalmente perché risiedono in un mezzo paragonabile ad un?interfaccia acqua-olio-acqua. Infatti, finora è stato impossibile determinare la struttura di questo complesso proteina-proteina, per questo abbiamo intrapreso la strada delle simulazioni. Bisogna quindi costruire il complesso con le due proteine, piazzarlo all?interno della membrana (medio oleoso) e finalmente aggiungere le molecole d?acqua e che circondano il tutto. Una volta che si hanno le posizioni (coordinate x, y, z) d?ogni atomo gli si assegnano dei parametri che determineranno le loro interazioni. Fatto questo, si risolve ?semplicemente? la legge di Newton (F=m?a). In questo modo conoscendo le forze ad un tempo T si possono calcolare le posizioni a tempo T+∆T. Risolvendo in modo iterativo si riesce ad avere una traiettoria temporale del sistema.
PI: Che cosa rende questo sistema tanto complesso?
SP: Le difficoltà sono molte, ma la principale è quella della grandezza del sistema. Per includere le due proteine, la membrana che le circonda e tutta l’acqua necessaria per simularle in condizioni realistiche, occorre impiegare circa 260.000 atomi. I sistemi standard stanno normalmente sotto i 100.000 atomi, e il “costo computazionale” di queste simulazioni cresce all’incirca con il cubo del numero d?atomi. La grandezza di questo sistema è fra le più grandi simulate con dinamica molecolare.
PI: Passiamo agli aspetti pratici. Come si realizza una simulazione del genere? Ci sono dei software specifici o dei linguaggi di programmazione orientati alla creazione di questi sistemi?
SP: Ci sono diversi software specifici per visualizzazione e manipolazione molecolare che permettono la costruzione modificazione di modelli strutturali di proteine. Poi ci sono dei pacchetti di software specifici che realizzano dinamica molecolare di proteine. Questi ultimi sono scritti generalmente in C o Fortran e sono ottimizzati per il calcolo parallelo: alcuni di questi scalano linearmente fino a centinaia di processori. Essi contengono interfacce grafiche che permettono di assegnare automaticamente i parametri d?interazione ai diversi tipi di atomi.
PI: Quanto tempo occorre per sviluppare tutto il modello di una simulazione di tale portata?
SP: Normalmente il set up di un sistema richiede circa un paio di giornate di lavoro. In questo caso, vista la dimensione e il tipo di simulazione, mi ci sono volute circa due settimane, perché risulta più complicato generare e controllare la corretta assegnazione di posizioni e parametri di ogni atomo. Basti dire che molti visualizzatori hanno un limite massimo di 100.000 atomi, che in questo caso è meno della metà del sistema. Questo ha richiesto anche ad un maggiore utilizzo di memoria e altre risorse.
PI: Che tempi richiederà la conclusione della simulazione?
SP: È difficile da dire perché non possiamo stimare a priori i tempi caratteristici dei movimenti delle proteine nel loro complesso. Per altri sistemi molto più piccoli, i tempi normali per simulare una decina di nano secondi (1×10-9) sono di circa due o tre mesi. Con questo sistema speriamo di completare lo studio in circa sei mesi, anche se non è escluso che sia necessario realizzare altre simulazioni di controllo.
PI: Ci sono altri istituti che portano avanti progetti simili al vostro?
SP: Sebbene ci sono tanti gruppi che si occupano di simulazioni biomolecolari, non ho notizie d?altri gruppi che si occupino di questo sistema in particolare. Penso che il motivo sia un poco dovuto alle risorse computazionali che il sistema richiede ma anche al fatto che la validazione dei risultati teorici richiede della stretta collaborazione fra gruppi teorici e sperimentali come si verifica all?interno del VIMM, e questo non è molto frequente.
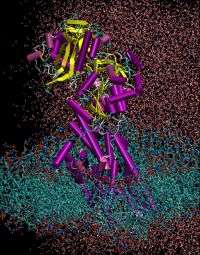 PI: Può illustrarci qualche particolare della simulazione con l’ausilio di alcuni screenshot?
PI: Può illustrarci qualche particolare della simulazione con l’ausilio di alcuni screenshot?
SP: Nella figura si può osservare una ?foto? del sistema. Per permettere un visione più chiara sono state cancellate le molecole d?acqua, che riempiono tutto il sistema. Ogni colore corrisponde ad un tipo di atomo diverso (rosso = ossigeno, bianco = idrogeno, ciano = carbonio, ecc.). La proteina è rappresentata come un ?cartoon?, cioè un disegno che segue lo scheletro e indica i diversi tipi dei motivi strutturali.
PI: Cosa vi ha spinto a preferire gli Xserve G5 rispetto alle altre soluzioni? Prestazioni, prezzo, disponibilità di software specifico?
SP: Essendo i programmi scritti in C e Fortran sono indipendenti dall?architettura (bastano dei buoni compilatori). Le prestazioni ottenute con questo software dai G5 sono leggermente superiori a quelle misurate con alcuni PC (tipo Xeon da 3 GHz), ma la vera motivazione di questa scelta e che l?Xserve G5 permette la creazione di un cluster facilmente espandibile con una gestione semplice in uno spazio ridotto e con un costo comparabile a quello necessario per l?acquisito di PC, che però sono meno facili da “parallelizzare”.
PI: Siete soddisfatti dei risultati che state ottenendo?
SP: Per ora siamo molto soddisfatti, il sistema operativo è molto stabile e le prestazioni sono molto alte, questo nonostante non si riesca ancora a sfruttare tutta la potenzialità del doppio processore. In realtà, per ottenere il meglio dal sistema, dobbiamo ancora ottimizzare al meglio la parallelizzazione del processo
PI: Qual è l’obiettivo finale della simulazione? Quali saranno i risvolti pratici che deriveranno da questo studio?
SP: Attualmente sono disponibili pochissime informazioni a livello strutturale (3D) riguardo questo complesso. Ci proponiamo di ottenere una struttura teorica del complesso SERCA-PLB, la quale sarà validata sperimentalmente dal gruppo di ?Calcium Signaling? del prof. Ernesto Carafoli presso il VIMM. Si spera che l?informazione derivata da questo studio possa fornire nuove indicazioni sul meccanismo d?azione di queste proteine e suggerire nuove strategie terapeutiche nella lotta contro le malattie cardiache.
PI: Avete intenzione di realizzare altre simulazioni con lo stesso sistema, o con sistemi simili?
SP: Sì, ci sono diversi progetti sempre nell?ambito delle simulazioni biomolecolari che interessano lo studio d?altre proteine di rilevanza biomedica. Speriamo per tanto di aggiungere presto altri nodi per aumentare la capacità di calcolo del cluster. Veniamo ora alla parte più tecnica del sistema, facendo qualche domanda a Pierpaolo Fantuzzi.
Punto Informatico: Signor Fantuzzi, ci può spiegare prima di tutto in cosa consiste il suo lavoro?
Pierpaolo Fantuzzi: Mi occupo dell’assistenza hardware e software su Mac in MacAT, un Apple Service che si occupa di soluzioni client-server con tecnologie Apple per la zona di Padova, e collaboro su BigRock nella stesura di articoli relativi ad Apple e MacOSX.
PI: E’ la prima volta che lavorate con il VIMM?
PF: Quella con il VIMM è una collaborazione continuativa che dura ormai da due anni. Personalmente mi occupo anche dell’assistenza generica alla rete informatica dell’istituto veneto di medicina molecolare (VIMM), il cui server è un “vecchio” Xserve G4. L’assistenza viene eseguita periodicamente due giorni alla settimana, e le problematiche più frequenti riguardano l’ottimizzazione della gestione della rete interna ed esterna.
PI: Come si realizza tecnicamente un cluster? Serve dell’hardware particolare o è sufficiente coordinare i diversi computer tramite appositi software?
PF: Esiste una sostanziale differenza quando si parla di calcolo distribuito via rete tra cluster e grid computing. Il primo è propriamente quello che nell’immaginario comune ci aspettiamo, ovvero un agglomerato di calcolatori con software e connessioni dedicate. Il secondo è un concetto più dinamico adatto a differenti situazioni: quest’ultimo è quello che abbiamo realizzato presso il VIMM di Padova. Sicuramente un hardware ottimizzato per questa tipologia di impiego garantisce prestazioni al vertice, ma spesso anche con hardware non espressamente dedicato è possibile realizzare sistemi in grado di competere con i più grandi supercomputer: un esempio di tutto ciò è la prima versione del cluster realizzato presso la Virginia University.
PI: Può darci qualche dettaglio sulle soluzioni hardware/software che avete impiegato per realizzare questo progetto?
PF: Quello che abbiamo fatto al VIMM è ottimizzare l’applicazione Amber (precedentemente compilata per MacOSX su PowerMac G5) per lavorare in parallelo su Xserve G5 e MacOSX, ricompilando ove possibile in MacMPI per sfruttare al meglio la parallelizzazione. L?intero sistema viene monitorato in remoto con i software AdminTools e Remote Desktop di Apple. L’hardware utilizzato si basa su 3 macchine Xserve G5 a doppio processore a 2 GHz con 2 GB di RAM per ogni server. I dischi utilizzati sono i Drive Module di Apple in tagli da 80 GB a 250 GB; la parte di rete è delegata ad uno switch Gigabit di HP. La versione del sistema operativo è MacOSX 10.3.5 Server con client illimitati per il controller e a 10 utenti per i nodi. I primi test sono stati eseguiti con xGrid di Apple e Pooch, mentre successivamente si è implementato LDAP e Open Directory per l’utilizzo di Amber.
PI: Quali difficoltà avete incontrato nella realizzazione del sistema?
PF: Le prime difficoltà che si incontrano sono logistiche, di assemblaggio e posizionamento in sede dei server e delle unità di storage. Anche il semplice cablaggio in rete necessita di un mimino studio, proporzionale alla dimensione del cluster da realizzare. Le difficoltà maggiori sono nell’ottimizzazione dei servizi LDAP, Open Directory, Netboot e di implementazione del software sulle varie unità di calcolo. Non tutte le applicazioni si adattano ad essere eseguite in parallelo su più macchine. Accanto alla fase di configurazione del cluster c’è anche un notevole sforzo nella ricompilazione del codice sorgente dell’applicazione mediante librerie Lam/Mpi o Mac/Mpi (dove possibile).
-PI: Che potenza di calcolo può raggiungere il sistema realizzato per il VIMM?
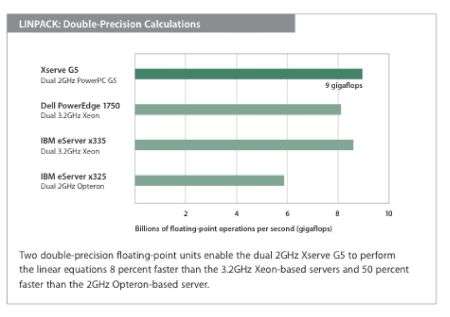
-PF: A causa di vincoli dettati dalla non totale compilazione MPI, attualmente il software non sfrutta appieno le potenzialità del doppio processore. In ogni caso abbiamo a disposizione un Xserve G5 e due Xserve Cluster Node con doppio processore a 2 GHz: per la tipologia di calcoli richiesti in questa simulazione, ogni singola unità può raggiungere una potenza di calcolo pari a nove gigaflops.
-PI: Quale vantaggio deriva, a suo parere, nell?utilizzare le soluzioni di Apple per realizzare cluster di questo tipo?
-PF: La prima è sicuramente la semplicità di assemblaggio e configurazione dei vari servizi. In seconda posizione vengono la robustezza di un sistema Unix (FreeBsd e Mach, MacOSX è un sistema dual-core) e il supporto da parte di Apple alle problematiche di sicurezza. Un ulteriore elemento positivo è il prezzo con cui Apple presenta i suoi server (Xserve, Cluster Node) e soluzioni per lo storage di grandi quantità di dati (xRaid). La possibilità di avere un sistema server (MacOSX Server) con client illimitati garantisce un risparmio non indifferente se paragonato ai concorrenti. Con MacOSX inoltre si ha la sicurezza di avere accesso a tecnologia standard di rete e di gestione anche in situazioni lavorative miste (Windows, Unix).
PI: MacOSX è a tutti gli effetti un sistema *nix: un altro grande protagonista di questo mondo è Linux. Cosa ci puoi dire di questo sistema e del mondo *nix in generale?
PF: Linux è un grande sistema, e può rappresentare una valida alternativa su piattaforma x86. L’installazione di Linux poi si è molto snellita in questi anni ed è praticabile, con qualche accorgimento, dalla maggior parte degli utenti. MacOSX resta comunque il sistema *nix più immediato e facile da usare. Se si vuole puntare su di una soluzione in cui hardware e software (di sistema ed applicazioni) risultino stabili ed integrate non si può che pensare ad Apple. Mi riferisco a tutti i software targati Apple, a partire dal sistema opertivo e passando per le allicazioni consumer, quelle professionali e quelle destinate al settore server (Final Cut Pro, Shake, Pro Logic, Xsan, Motion, Apple Remote Destop, Qmaster, Compressor, ecc.).
PI: Possiamo visionare qualche dettaglio del cluster con l’ausilio di alcune fotografie?
PF: Nella prima fotografia abbiamo una vista frontale del sistema di Xserve e le unità cluster node G5. Di seguito un Xserve in modalità manutenzione, e un dettaglio sul retro delle macchine dove sono ben visibili le porte di rete.

Ringraziamo il Dott. Sergio Pantano, responsabile del progetto presso il VIMM, e il Sig. Pierpaolo Fantuzzi, tecnico hardware-software di MacAT, per la disponibilità accordataci e per la collaborazione nelle realizzazione di questo articolo.
Domenico Galimberti
Ti potrebbe interessare





18 feb 2005